TREC 2004 Genomics Track Final Protocol
William Hersh
Oregon Health & Science University, Track Chair
hersh@ohsu.edu
Last updated - February 23, 2005
This file contains the final protocol for the TREC 2004 Genomics
Track. After the regular track cycle was completed, the data for
the categorization task was updated to reflect additional changes to
the Mouse Genomic Informatics (MGI) database that had been done while
the track was taking place. This was done to provide the most
up-to-date data to those continuing to perform experiments with track
data. An update file describes the
changes to the data.
A total of 33 groups participated in the 2004 Genomics Track,
making it
the track with the most participants in all of TREC 2004. A toal
of 145 runs were submitted. For the ad hoc task, there were 47
runs from 27 groups, while for the categorization task, there
were 98
runs from 20 groups. The runs
of the categorization task were distributed across the subtasks as
follows: 59 for the
triage subtask, 36 for the annotation hierarchy subtask, and three for
the
annotation hierarchy plus evidence code subtask.
The data will be available to
non-TREC participants in early 2005. A listing
of the available data is available.
Introduction
This document contains the final protocol for the TREC
2004 Genomics Track.
This protocol had the same general structure as the 2003
track, in that there was a basic information retrieval task and a
more
experimental text categorization task. We decided, however,
to not use the words "primary" and "secondary" as we did in 2003 to
describe the two
tasks. Instead, we called the two tasks "ad hoc retrieval" and
"categorization." The ad hoc retrieval task focused on
"regular" document retrieval, while the categorization task focused
on classifying documents containing "experimental evidence" allowing
assignment of GO codes.
The roadmap
for the Genomics Track called for modifying one experimental "facet"
each year. For the purposes of the roadmap (based on the NSF
grant proposal), last year (2003) was Year 0. This means that
2004 was Year 1. The original plan was to add new
types of content in Year 1 and new types of information needs in Year
2. Because we were unable to secure substantial numbers of
full text documents for the ad hoc retrieval task in 2004, we decided
to reverse the order of the roadmap for Years 1 and 2.
This means we focused on new types of information needs for 2004.
There were some resources we made extensive use of in 2004 for the
categorization task:
- The Gene Ontology (GO)
- a controlled terminology used to annotate the function of genes and
proteins.
- Mouse Genome
Informatics (MGI) - the model organism database for the mouse,
which, among other things, links literature references to gene function
through the use of GO codes.
It is important to have a general understanding of these resources,
including the codes
used to assign the evidence supporting an annotation using GO.
Here are some pointers to get started:
Ad Hoc Retrieval Task
The structure of this task was a conventional searching task based on a
10-year subset of MEDLINE (about 4.5 million documents and 9 gigabytes
in size) and 50 topics dervied from information needs obtained via
interviews of real biomedical researchers. There was no training
data, although sample topics and relevance judgments were
available.
Documents
The document collection for this task was a 10-year subset of the MEDLINE
bibliographic database of the biomedical literature. MEDLINE can
be searched by anyone in the world using the PubMed system of the National Library of Medicine (NLM),
which maintains both MEDLINE and PubMed. The full MEDLINE
database contains over 13 million references dating back to 1966 and is
updated on a daily basis.
The subset of MEDLINE used for the TREC 2004 Genomics Track
consisted of 10 years of completed citations from the database
inclusive
from 1994 to 2003. Records were extracted using the Date
Completed (DCOM) field for all references in the range of 19940101 -
20031231. This provided a total of 4,591,008
records, which is about one third of the full MEDLINE database. A
gzipped list of the PubMed IDs (PMIDs) is available (10.6 megabytes
compressed).
We used the DCOM field and not the Date Published (DP). DCOM and
DP are not linked in any formal manner. There are plenty of cases
where something is not indexed until quite long after it was
published. This may be due to several reasons including the
indexer backlog, a new interest in indexing a particular journal -
where they have to go back and index the collection,
etc.. So this is not an inconsistency or problem with the data.
We pulled the collection using the DCOM field and did not worry about
when the articles were published, last revised (LR), or created
(DA). We had to pick a date to use to define the boundaries for
the 10 years of MEDLINE and
the DCOM field was chosen with the thought that it would provide us
with a good indicator. The numbers for the complete 4,591,008
collection are as follows:
- 2,814 ( 0.06%) DPs prior to 1980
- 8,388 ( 0.18%) DPs prior to 1990
- 138,384 ( 3.01%) DPs prior to 1994
The remaining 4,452,624 (96.99%) DPs were within the 10 year
period of 1994-2004.
The data includes all of the PubMed fields identified in the MEDLINE
Baseline record and only the PubMed-centric tags are removed from the
XML version. A description of the various fields of MEDLINE are
available at:
http://www.ncbi.nlm.nih.gov/entrez/query/static/help/pmhelp.html#MEDLINEDisplayFormat
The only field at that link not in the subset is the Entrez Date (EDAT)
field because that information is not included in the MEDLINE Baseline
record. It should also be noted that not all MEDLINE records have
abstracts, usually because the article itself does not have an
abstract. In general, about 75% of MEDLINE records have
abstracts. In our subset, there are 1,209,243 (26.3%) records
without abstracts.
The MEDLINE subset is available in two formats:
- MEDLINE - ASCII text with fields indicated and delimited by 2-4
character abbreviations (uncompressed - 9,587,370,116 bytes, gzipped -
2,797,589,659 bytes)
- XML - NLM XML format (uncompressed - 20,567,278,551 bytes,
gzipped - 3,030,576,659 bytes)
Groups contemplating using their own MEDLINE collection or filtering
from "live" PubMed should be aware of the following caveats (from Jim
Mork of NLM):
- Some citations may differ due to revisions or corrections.
- Some citations may no longer exist. The bulk of the
citations come from the 2004 Baseline; some may have been removed since
that baseline was created.
- UTF-8 characters have been translated to 7-bit ASCII whereas the
live PubMed system provides UTF-8 data and in some cases puts in
characters like the inverted question mark.
- The XML and ASCII files have been modified to conform to the
MEDLINE Baseline format. This is a subtle difference but one that
may cause changes in the files.
Topics
The ad hoc retrieval task consisted of 50 topics derived from
interviews
eliciting information needs of real biologists. We collected a
total of 74 information needs statements. These were then honed
into topics that represent reasonable kinds of information needs for
which a biologist might search the literature and that retrieve a
reasonable (i.e., not zero and not thousands) number of relevant
documents. The topics are formatted in XML and have the
following fields:
- ID - 1 to 50
- Title - abbreviated statement of information need
- Information need - full statement information need
- Context - background information to place information need in
context
We also created an additional five sample topics to demonstrate what
the topics looked like before the official topics were available.
These did not have relevance judgments.
Relevance judgments
Relevance judgments were done using the conventional "pooling
method" whereby a fixed number of top-ranking documents (depending on
the total number of documents and available resources for judging) from
each offiicial run are pooled and provided to an individual (blinded to
the query statement and participant who they came from) who judges them
as definitely relevant (DR), possibly relevant (PR), or not relevant
(NR) to the topic. A set of documents were also judged in
duplicate to assess interjudge reliability. For
the standpoint of the official results, which require binary relevance
judgments, documents that were rated
DR or PR were considered relevant.
The judgments were done by two individuals with backgrounds in
biology. The pools were built from the top-precedence run
from each of the 27 groups. We took the top 75 documents for each
topic and eliminated the duplicates to create a single pool for
each. The average pool size (average number of documents judged
per topic) was 976, with a range of 476-1450. The table below
shows the total number
of judgments and their distribution for the 50 topics.
There
are two files of relevance judgments:
04.judgments.txt - a
file of all relevance judgments done, based on the format:
<topic><PMID><judgment>
where
<judgment> = 1 (DR), 2 (PR), or 3 (NR)
04.qrels.txt - a
file of relevant documents, based on the format:
<topic><0><PMID><judgment>
The
qrels file is used for input to the trec_eval program to generate
recall-precision results. The trec_eval
program considers any document with a judgment greater than 0 to be
relevant,
which in the trecgen04-qrels.txt file is all documents.
Those wanting to experiment with DR-only
relevance judgments can do so, although only 47 topics can be used
(since 3
have no DR documents).
| Topic |
Total Judgments
|
Definitely relevant |
Possibly relevant |
Not relevant |
Definitely and probably relevant |
| 1 |
879 |
38 |
41 |
800 |
79 |
| 2 |
1264 |
40 |
61 |
1163 |
101 |
| 3 |
1189 |
149 |
32 |
1008 |
181 |
| 4 |
1170 |
12 |
18 |
1140 |
30 |
| 5 |
1171 |
5 |
19 |
1147 |
24 |
| 6 |
787 |
41 |
53 |
693 |
94 |
| 7 |
730 |
56 |
59 |
615 |
115 |
| 8 |
938 |
76 |
85 |
777 |
161 |
| 9 |
593 |
103 |
12 |
478 |
115 |
| 10 |
1126 |
3 |
1 |
1122 |
4 |
| 11 |
742 |
87 |
24 |
631 |
111 |
| 12 |
810 |
166 |
90 |
554 |
256 |
| 13 |
1118 |
5 |
19 |
1094 |
24 |
| 14 |
948 |
13 |
8 |
927 |
21 |
| 15 |
1111 |
50 |
40 |
1021 |
90 |
| 16 |
1078 |
94 |
53 |
931 |
147 |
| 17 |
1150 |
2 |
1 |
1147 |
3 |
| 18 |
1392 |
0 |
1 |
1391 |
1 |
| 19 |
1135 |
0 |
1 |
1134 |
1 |
| 20 |
814 |
55 |
61 |
698 |
116 |
| 21 |
676 |
26 |
54 |
596 |
80 |
| 22 |
1085 |
125 |
85 |
875 |
210 |
| 23 |
915 |
137 |
21 |
757 |
158 |
| 24 |
952 |
7 |
19 |
926 |
26 |
| 25 |
1142 |
6 |
26 |
1110 |
32 |
| 26 |
792 |
35 |
12 |
745 |
47 |
| 27 |
755 |
19 |
10 |
726 |
29 |
| 28 |
836 |
6 |
7 |
823 |
13 |
| 29 |
756 |
33 |
10 |
713 |
43 |
| 30 |
1082 |
101 |
64 |
917 |
165 |
| 31 |
877 |
0 |
138 |
739 |
138 |
| 32 |
1107 |
441 |
55 |
611 |
496 |
| 33 |
812 |
30 |
34 |
748 |
64 |
| 34 |
778 |
1 |
30 |
747 |
31 |
| 35 |
717 |
253 |
18 |
446 |
271 |
| 36 |
676 |
164 |
90 |
422 |
254 |
| 37 |
476 |
138 |
11 |
327 |
149 |
| 38 |
1165 |
334 |
89 |
742 |
423 |
| 39 |
1350 |
146 |
171 |
1033 |
317 |
| 40 |
1168 |
134 |
143 |
891 |
277 |
| 41 |
880 |
333 |
249 |
298 |
582 |
| 42 |
1005 |
191 |
506 |
308 |
697 |
| 43 |
739 |
25 |
170 |
544 |
195 |
| 44 |
1224 |
485 |
164 |
575 |
649 |
| 45 |
1139 |
108 |
48 |
983 |
156 |
| 46 |
742 |
111 |
86 |
545 |
197 |
| 47 |
1450 |
81 |
284 |
1085 |
365 |
| 48 |
1121 |
53 |
102 |
966 |
155 |
| 49 |
1100 |
32 |
41 |
1027 |
73 |
| 50 |
1091 |
79 |
223 |
789 |
302 |
| Total |
48753 |
4629 |
3639 |
40485 |
8268 |
As noted above, we performed some overlapping judgments to assess
interjudge consistency using Cohen's kappa measure. The table
below shows the results of the overlapping judgments. The value
obtained for Cohen's kappa was 0.51, indicating "fair" consistency.
Judge 1
/ Judge 2
|
Definitely relevant
|
Possibly relevant
|
Not relevant
|
Total
|
Definitely relevant
|
62
|
35
|
8
|
105
|
Possibly relevant
|
11
|
11
|
5
|
27
|
Not relevant
|
14
|
57
|
456
|
527
|
Total
|
87
|
103
|
469
|
659
|
Results
Recall and precision for the ad hoc retrieval task were calculated in
the classic IR way, using the preferred TREC statistic of mean average
precision (average precision at each point a relevant document is
retrieved, also called MAP). This was done using the standard
TREC
approach of participants submitting their results in the format for
input to Chris Buckley’s trec_eval program. The code for
trec_eval is available at ftp://ftp.cs.cornell.edu/pub/smart/
. There are several versions of trec_eval, which differ mainly in
the statistics they calculate in their output. We recommend the
following version of trec_eval, which should compile with any C
compiler:
ftp://ftp.cs.cornell.edu/pub/smart/trec_eval.v3beta.shar .
Please note a few "quirks" about trec_eval:
- It uses some Unix-specific system calls so would require
considerable modification to run on another platform.
- The aggregate statistics that are presented at the end of the
file (averages for precision at points of recall, average precision,
etc.) only include queries for which one or more documents were
retrieved. Therefore, you should insert a "dummy" document in
your output for queries that retrieve no real documents so that your
aggregate scores are averaged over all 50 documents.
The trec_eval program requires two files for input. One file is
the topic-document output, sorted by each topic and then subsorted by
the order of the IR system output for a given topic. This
format is required for official runs submitted to NIST to obtain
official scoring.
The topic-document ouptut should be formatted as follows:
1 Q0 12474524 1 5567 tag1
1 Q0 12513833 2 5543 tag1
1 Q0 12517948 3 5000 tag1
1 Q0 12531694 4 2743 tag1
1 Q0 12545156 5 1456 tag1
2 Q0 12101238 1 3.0 tag1
2 Q0 12527917 2 2.7 tag1
3 Q0 11731410 1 .004 tag1
3 Q0 11861293 2 .0003 tag1
3 Q0 11861295 3 .0000001 tag1
where:
- The first column is the topic number (1-50).
- The second column is the query number within that topic.
This is currently unused and must always be Q0.
- The third column is the official PubMedID of the retrieved
document.
- The fourth column is the rank the document is retrieved
- The fifth column shows the score (integer or floating point) that
generated the ranking. This score MUST be in descending
(non-increasing) order. The trec_eval program ranks documents
based on the scores, not the ranks in column 4. If a submitter
wants the exact ranking submitted to be evaluated, then the SCORES must
reflect that ranking.
- The sixth column is called the "run tag" and must be a unique
identifier across all runs submitted to TREC. Thus, each run tag
should have a part that identifies the group and a part that
distinguishes runs from that group. Tags are restricted to 12 or
fewer letters and numbers, and *NO* punctuation, to facilitate labeling
graphs and such with the tags.
The second file required for trec_eval is the relevance judgments,
which are called "qrels" in TREC jargon. More information about
qrels can be found at http://trec.nist.gov/data/qrels_eng/
. The qrels file is in the following format:
1 0 12474524 1
1 0 12513833 1
1 0 12517948 1
1 0 12531694 1
1 0 12545156 1
2 0 12101238 1
2 0 12527917 1
3 0 11731410 1
3 0 11861293 1
3 0 11861295 1
3 0 12080468 1
3 0 12091359 1
3 0 12127395 1
3 0 12203785 1
where:
- The first column is the topic number (1-50).
- The second column is always 0.
- The third column is the PubMedID of the document.
- The fourth column is always 1.
Experiments
Each group was
allowed to submit up to two official runs. Each was classified
into one of the following two categories:
- Automatic - no system or query tuning for the specific topics
(systems can be tuned for the sample queries without calling the runs
manual)
- Manual - with system or query tuning for the specific topics
Categorization Task
A major activity of most model organism database projects is to assign
codes from the Gene Ontology (GO) to annotate the function of genes and
proteins. The GO consists of three structured, controlled
vocabularies (ontologies) that describe gene products in terms of their
associated (1) biological processes, (2) cellular components, and (3)
molecular functions. Each assigned GO code is also designated
with a level of evidence indicating the specific experimental evidence
in support of assignment of the code. For more information on
these, visit the GO Web site
.
In the categorization task, we attempted to mimic two of the
classification
acitivites carried out by human annotators in the mouse genome
informatics (MGI) system: a triage
task and two variants of MGI's annotation
task. Systems were required to classify full-text
documents
from a two-year span (2002-2003) of three journals. The first
year's (2002) documents comprised the training data, while the second
year's (2003) documents made up the test data.
Like most tracks within TREC, the Genomics Track operates on the
honor system whereby participants follow rules to insure that
inappropriate data is not used and results are comparable across
systems. In this task, many of the classification answers are
obtainable by searching MGI. In addition, data for the annotation
task can be used to infer answers for the triage task. It is
therefore imperative that users not make inappropriate use of
MGI or other data, especially for the test data.
Background
One of the goals of MGI is to provide structured, coded
annotation of gene function from the biological literature.
Human curators identify genes and assign GO codes about gene
function with another code describing the experimental evidence for the
GO code. The huge amount of literature to curate
creates a challenge for MGI, as their resources are not unlimited.
As such, they employ a three-step process to identify the papers
most likely to describe gene function:
- About mouse - The first step is to identify articles about mouse
genomics biology. Articles from several hundred
journals are searched for the words mouse,
mice, or murine. Articles
caught in this "mouse trap" are further analyzed for inclusion in
MGI. At present, articles are search in a Web browser one at a
time because full-text searching is not available for all of the
journals.
- Triage - The second step is to determine whether the identified
articles should be sent for curation. MGI curates
articles not only for GO terms, but also for other aspects of biology,
such as gene mapping, gene expression data, phenotype description, and
more. The goal of this triage process is to limit
the number of articles sent to human curators for more exhaustive
analysis. Articles that pass this step go into the
MGI system with a tag for GO, mapping, expression, etc.. The
rest of the articles do not go into MGI. Our triage task involved
correctly classifying which documents have been selected for GO
annotation in this process.
- Annotation - The third step is the actual curation with GO terms.
Curators identify genes for which there is experimental
evidence to warrant assignment of GO codes. Those
GO codes are assigned, along with a code for each indicating the type
of evidence. There can more than one gene assigned GO codes in a
given paper and there can be more than one GO code assigned to a
gene. In general, and in our collection, there is only one
evidence code per GO code assignement per paper. Our annotation
task involved a modification of this annotation step as described below.
Documents
The documents for the tasks consisted of articles from three journals
over two years, reflecting the full-text data we were able to
obtain from Highwire Press.
The journals available were Journal
of Biological Chemistry (JBC), Journal
of Cell Biology (JCB), and Proceedings
of the National Academy of Science (PNAS). These
journals have a good proportion of mouse genome articles. Each
of the papers from these journals was in SGML format.
Highwire's DTD
and its documentation are available. We
designated 2002 articles as training data and 2003 articles as test
data. The documents for the tasks came from a subset of these
articles that get caught by the "mouse trap" described above. A
crosswalk or look-up table was provided that matched an identifier for
each Highwire article (its file name) and its corresponding PubMed ID
(PMID). The table below shows the total number of articles and
the number in the subset the track used.
| Journal |
2002 papers - total, subset
|
2003 papers - total, subset
|
Total papers - total, subset
|
JBC
|
6566, 4199
|
6593, 4282
|
13159, 8481
|
JCB
|
530, 256
|
715, 359
|
1245, 615
|
PNAS
|
3041, 1382
|
2888, 1402
|
5929, 2784
|
Total papers
|
10137, 5837
|
10196, 6043
|
20333, 11880
|
The following table lists the the files containing the documents and
related data.
File contents
|
Training data file name
|
Test data file name
|
Full-text document collection in
Highwire SGML format
|
train.tar.Z |
test.tar.Z |
| Crosswalk files of PMID,
Highwire file name of article,
journal code, and year of paper publication |
train.crosswalk.txt |
test.crosswalk.txt |
Positive training examples
|
triage+train.txt |
triage+test.txt |
The SGML training document collection is 150 megabytes in size
compressed and 449 megabytes uncompressed. The SGML test document
collection is 140 megabytes compressed and 397 megabytes uncompressed.
Many gene names have Greek or other
non-English characters, which can present a problem for those
attempting
to recognize gene names in the text. The Highwire SGML appears to
obey the rules posted on the NLM Web site with regards to these
characters (http://www.ncbi.nlm.nih.gov/entrez/query/static/entities.html).
Triage Task
The goal of this task was to correctly identify which papers were
deemed to have experimental evidence warranting annotation with GO
codes. Positive examples were papers designated for GO
annotation by MGI. However, some of these papers
had not yet been annotated. Negative examples were all papers
not designated for GO annotation in the operational MGI system.
For the training data (2002), there were 375 positive examples, meaning
that there were 5837-375 = 5462 negative examples. For the test
data (2003), there were 420 positive examples, meaning that there were
6043-420 = 5623 negative examples.
Annotation Task
The primary goal of this task was to correctly identify, given the
article and gene name, which of the GO hierarchies (also called
domains) have terms within them that have been annotated. Note
that the goal of this task was not to select the correct GO term, but
rather to select the one or more GO hierarchies (biological process,
cellular component, and molecular function, also called domains) from
which terms had been
selected to annotate the gene for the article. Papers which had
been annotated had from one to three hierarchies. There were
328 papers in the collection that had GO terms assigned.
We also included a batch of 558 papers that had a gene name
assigned but were used for other purposes by MGI. As such, these
papers had no GO annotations. These papers did, however, have one
or more gene assigned for the purposes.
A secondary goal of this task was to identify the correct GO evidence
code that goes along with the hierarchy code.
Task Data
The figure below shows where the positive and negative examples for the
test and training came from in the MGI system.
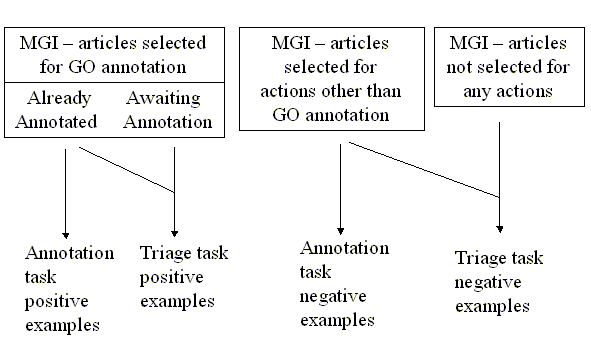
Triage task data
The following files contain the positive examples (negative examples
can be obtained by subtracting these documents from the crosswalk files
above):
- Text file of positive examples for training data
- Text file of positive examples for test data
Annotation task data
The table below shows the contents, names, and line counts of the data
files for this task. Here is an interpretation of the numbers in
the table: For the training data, there are a total of 504
documents that are either positive (one or more GO terms assigned) and
negative (no GO terms assigned) examples. From these documents, a
total of 1291 genes have been assigned by MGI. (The file
gtrain.txt containts the MGI identifier, the gene symbol, and the gene
name. It does not contain any other synonyms.) There are
1418 unique document-gene pairs in the training data. The data
from the first three rows of the table differ from the rest in that
they contain data merged from positive and negative examples.
These are what would be used as input for systems to nominate GO
domains or the GO domains plus their evidence codes per the annotation
task. When the test data are released, these three files are the
only ones that will be provided.
For the positive examples in the training data, there are 178 documents
and 346 document-gene pairs. There are 589 document-gene name-GO
domain tuples (out of a possible 346 * 3 = 1038). There are 640
document-gene name-GO domain-evidence code tuples. A total of 872
GO plus evidence codes have been assigned to these documents.
For the negative examples, there are 326 documents and 1072
document-gene pairs. This means that systems could possibly
assign 1072*3 = 3216 document-gene name-GO domain tuples.
File contents
|
Training data file name
|
Training data count
|
Test data file name
|
Test data count
|
Documents - PMIDs
|
ptrain.txt
|
504
|
ptest.txt
|
378
|
Genes - Gene symbol, MGI
identifier, and gene name for all used
|
gtrain.txt
|
1294
|
gtest.txt
|
777
|
| Document gene pairs - PMID-gene
pairs |
pgtrain.txt |
1418 |
pgtest.txt |
877 |
Positive examples - PMIDs
|
p+train.txt
|
178
|
p+test.txt
|
149
|
Positive examples - PMID-gene
pairs
|
pg+train.txt
|
346
|
pg+test.txt
|
295
|
Positive examples -
PMID-gene-domain tuples
|
pgd+train.txt
|
589
|
pgd+test.txt
|
495
|
Positive examples -
PMID-gene-domain-evidence tuples
|
pgde+train.txt
|
640
|
pgde+test.txt
|
522
|
Positive examples - all
PMID-gene-GO-evidence tuples
|
all+train.txt
|
872
|
all+test.txt
|
693
|
Negative examples - PMIDs
|
p-train.txt
|
326
|
p-test.txt
|
229
|
Negative examples - PMID-gene
pairs
|
pg-train.txt
|
1072
|
pg-test.txt
|
582
|
The next table shows the actual data in each of the above files.
The data are in ASCII format. The field names in are delimited
below with vertical bars, but in the files, the field contents are
delimited with tab characters.
Training data file name
|
Data in file
|
ptrain.txt
|
PMID
|
gtrain.txt
|
Gene symbol|MGI ID|Gene name
|
| pgtrain.txt |
PMID|Gene symbol
|
p+train.txt
|
PMID
|
pg+train.txt
|
PMID|Gene symbol
|
pgd+train.txt
|
PMID|Gene symbol|GO domain |
pgde+train.txt
|
PMID|Gene symbol|GO domain|GO
evidence code |
all+train.txt
|
PMID|MGI ID|Gene symbol|Gene
name|GO domain|GO code|GO name|GO evidence code |
p-train.txt
|
PMID |
pg-train.txt
|
PMID|Gene symbol |
Examples
Some examples from the data may make the above more clear. Let's
start with a positive example and consider the following paper:
Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C,
Feldman GM, Nishikomori R, O'Shea JJ. STAT4 serine
phosphorylation is critical for IL-12-induced IFN-gamma production but
not for cell proliferation. Proc
Natl Acad Sci U S A. 2002 Sep 17;99(19):12281-6. Epub 2002 Sep
04. PMID: 12213961 [PubMed - indexed for MEDLINE].
The PMID for this paper is 12213961. Using grep, we can look at
the instances of this documents in the various files (long lines
truncated):
$ grep 12213961 *.txt
all+train.txt:12213961 MGI:107776 Gadd45b growth arrest and DNA-damage-inA
all+train.txt:12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inA
all+train.txt:12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inS
all+train.txt:12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inS
all+train.txt:12213961 MGI:1346870 Map2k6 mitogen activated protein kinasA
all+train.txt:12213961 MGI:103062 Stat4 signal transducer and activatorA
all+train.txt:12213961 MGI:103062 Stat4 signal transducer and activatorA
all+train.txt:12213961 MGI:103062 Stat4 signal transducer and activatorA
all+train.txt:12213961 MGI:103062 Stat4 signal transducer and activatorA
p+train.txt:12213961
pg+train.txt:12213961 Gadd45b
pg+train.txt:12213961 Gadd45g
pg+train.txt:12213961 Map2k6
pg+train.txt:12213961 Stat4
pgd+train.txt:12213961 Gadd45b BP
pgd+train.txt:12213961 Gadd45g BP
pgd+train.txt:12213961 Map2k6 BP
pgd+train.txt:12213961 Stat4 MF
pgd+train.txt:12213961 Stat4 CC
pgd+train.txt:12213961 Stat4 BP
pgde+train.txt:12213961 Gadd45b BP IDA
pgde+train.txt:12213961 Gadd45g BP IDA
pgde+train.txt:12213961 Gadd45g BP TAS
pgde+train.txt:12213961 Map2k6 BP IDA
pgde+train.txt:12213961 Stat4 MF IDA
pgde+train.txt:12213961 Stat4 CC IDA
pgde+train.txt:12213961 Stat4 BP IDA
pgtrain.txt:12213961 Gadd45b
pgtrain.txt:12213961 Gadd45g
pgtrain.txt:12213961 Map2k6
pgtrain.txt:12213961 Stat4
ptrain.txt:12213961
train.crosswalk.txt:pq1902012281.gml 12213961 PNAS 2002
triage+train.txt:12213961
From the train.crosswalk.txt file, we can find the full-text Highwire
SGML version of the paper, which has the file name pq1902012281.gml
(and is in train.tar.Z). This paper has GO codes, so is a
positive example for the triage task and appears in the file
triage+train.txt. It also is a positive example for the
annotation task, so appears in the file of all annotation training
papers, ptrain.txt, as well as the file of positive examples,
p+train.txt. This paper is annotated with four genes:
Gadd45b, Gadd45g, Map2k6, and Stat4. These genes appear with the
paper in the files pgtrain.txt and pg+train.txt.
From the file all+train.txt, we can get all of the GO and evidence code
assignments for these genes in this paper. There are a total of
nine assignments:
12213961 MGI:107776 Gadd45b growth arrest and DNA-damage-inducible 45 beta BP GO:0000186 activation of MAPKK IDA
12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inducible 45 gamma BP GO:0000186 activation of MAPKK IDA
12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inducible 45 gamma BP GO:0042095 interferon-gamma biosynthesis TAS
12213961 MGI:1346325 Gadd45g growth arrest and DNA-damage-inducible 45 gamma BP GO:0045063 T-helper 1 cell differentiation TAS
12213961 MGI:1346870 Map2k6 mitogen activated protein kinase kinase 6 BP GO:0006468 protein amino acid phosphorylation IDA
12213961 MGI:103062 Stat4 signal transducer and activator of transcription 4 MF GO:0003677 DNA binding IDA
12213961 MGI:103062 Stat4 signal transducer and activator of transcription 4 CC GO:0005634 nucleus IDA
12213961 MGI:103062 Stat4 signal transducer and activator of transcription 4 BP GO:0008283 cell proliferation IDA
12213961 MGI:103062 Stat4 signal transducer and activator of transcription 4 BP GO:0019221 cytokine and chemokine mediated signaling pathway IDA
Gadd45b has one assignment, and as such, appears in the files
pg+train.txt, pgd+train.txt, and pgde+train.txt only once.
Gadd45g has three GO assignments from two different codes in the same
hierarchy. It thus appears once in pg+train.txt, once in
pgd+train.txt, and twice in pgde+train.txt. The latter situation
occurs because even though the two GO codes are in the same hierarchy,
they have different evidence codes associated with them. Map2k6
is like Gadd45b, with one assignment and occuring once in the
appropriate files. Stat4 has four GO assignments that include all
three GO hierarchies. As such, it appears once in pg+train.txt,
three times in pgd+train.txt, and three times in pgde+train.txt.
Now let's consider a negative example based on the following paper:
Vivian JL, Chen Y, Yee D, Schneider E, Magnuson T. An allelic
series of mutations in Smad2 and Smad4 identified in a genotype-based
screen of N-ethyl-N- nitrosourea-mutagenized mouse embryonic stem
cells. Proc Natl Acad Sci U S A.
2002 Nov 26;99(24):15542-7. Epub 2002 Nov 13. PMID: 12432092
[PubMed - indexed for MEDLINE]
This paper has PMID 12432092, so we use grep as follows:
$ grep 12432092 *.txt
p-train.txt:12432092
pg-train.txt:12432092 Smad2
pg-train.txt:12432092 Smad4
pgtrain.txt:12432092 Smad2
pgtrain.txt:12432092 Smad4
ptrain.txt:12432092
train.crosswalk.txt:pq2402015542.gml 12432092 PNAS 2002
As can be seen, negative examples are simpler. From the PMID
12432092, we can use the train.crosswalk.txt file to find the full-text
Highwire SGML version of the paper, which has the file name
pq2402015542.gml (and is also in train.tar.Z). Because the
paper is in the training data set, it appears in ptrain.txt.
Because it is a negative example, it appears in p-train.txt. The
paper does have two genes associated (by MGI) with it, Smad2 and
Smad4. These appear in the files of overall article-gene pairs
and negative examples of article-gene pairs.
Gene tagging tools
Several research groups graciously allowed their open-source tools for
gene tagging to be used for non-commercial purposes
on an "as is" basis. These included:
More information about each can be found on their respective Web
sites. Alex Morgan of MITRE also maintains a page of BioNLP tools:
http://www.tufts.edu/~amorga02/bcresources.html
Evaluation measures
The framework for evaluation in the categorization task was based on
the
following table of possibilities:
|
Relevant (classified)
|
Not relevant (not classified)
|
Total
|
Retrieved
|
True positive (TP)
|
False positive (FP)
|
All retrieved (AR)
|
Not retrieved
|
False negative (FN)
|
True negative (TN)
|
All not retrieved (ANR)
|
|
All positive (AP)
|
All negative (AN)
|
|
The consensus of the track was to use a utility measure for the triage
task and F measure for the annotation tasks. We developed a
Python program that calculated both statistics for each
task.
Triage evaluation measures
The measure for the triage task was the utility measure often applied
in
text categorization research and used by the former TREC Filtering
Track. This measure contains coefficients for the
utility of retrieving a relevant and retrieving a nonrelevant document.
We used a version that was normalized by the best possible
score:
Unorm = Uraw / Umax
For a test collection of documents to categorize, Uraw was
calculated as follows:
Uraw = (ur * TP)
+ (unr * FP)
where:
- ur = relative utility of relevant document
- unr = relative utility of nonrelevant document
We used values for ur and unr that are driven by
boundary cases for different results. In
particular, we wanted the measure to have the following characteristics:
- Completely perfect prediction - Unorm = 1
- All documents designated positive (triage everything) - 1 > Unorm
> 0
- All documents designated negative (triage nothing) - Unorm
= 0
- Completely imperfect prediction - Unorm < 0
In order to achieve the above boundary cases, we had to set ur
> 1. The ideal approach would have been to interview MGI
curators and
use decision-theoretic approaches to determine their utilty.
However, time did not permit us to do that. Deciding that the
triage-everything approach should have a higher score than the
triage-nothing approach, we estimated that a Unorm in the
range of 0.25-0.3 for the triage-everything condition would be
appropriate. Solving for the above boundary cases with Unorm
~ 0.25-0.3 for that case, we obtained a value for ur ~
20. To keep calculations simple, we chose a value of ur
= 20. The table below shows the value of Unorm for the
boundary cases.
Situation
|
Unorm - Training |
Unorm - Test |
| Completely perfect prediction |
1.0
|
1.0
|
| Triage everything |
0.27
|
0.33
|
| Triage nothing |
0
|
0
|
| Completely imperfect prediction |
-0.73
|
-0.67
|
The measure Umax is calculated by assuming all relevant
documents are retrieved and no nonrelevant documents are retrieved:
Umax = ur * AP
(This happens to equal AN.)
Thus, for the training data,
Uraw = (20 * TP) - FP
Umax = 20 * 375 = 7500
Unorm = [(20 * TP) - FP] /
7500
(If you plug in the bounary conditions to the these equations, you
should obtain the results specificed above.)
Likewise, for the test data,
Uraw = (20 * TP) - FP
Umax = 20 * 420 = 8400
Unorm = [(20 * TP) - FP] /
8400
Annotation evaluation measures
The measures for the annotation subtasks were based on the notion of
identifying tuples of data. Given the article and gene, systems
designated one or both of the folloing tuples:
- <article, gene, GO hierarchy code>
- <article, gene, GO hierarchy code, evidence code>
We employed a global recall, precision, and F measure evaluation
measure
for each subtask:
Recall = number of tuples correctly
identified / number of correct tuples = TP / AP
Precision = number of tuples correctly
identified / number of tuples identified = TP / AR
F = (2 * recall * precision) / (recall + precision)
For the training data, the number of correct <article, gene, GO
hierarchy code> tuples was 593, while the number of correct
<article, gene, GO hierarchy code, evidence code> tuples for the
test data was 644.
Submitting results
We developed a Python program that calculates the normalized utility,
recall, precision, and F score for all of the subtask
submissions. The general format of the files is tab-delimited
ASCII, with the first column denoting the subtask (triage, annotation
hierarchy, or annotation hierarchy plus evidence code) and the last
column containing a run tag, unique for the participant and the
run. Below are the formats followed by an example for the
specific subtasks:
Triage
triage <PMID> tag
triage 12213961 tag
Annotation hierarchy
annhi <PMID> <Gene> <Hierarchy code> tag
annhi 12213961 Stat4 BP tag
Annotation hierachy plus evidence
annhiev <PMID> <Gene> <Hierarchy code> <Evidence code> tag
annhiev 12213961 Stat4 BP IDA tag
(Submitted results should not include a header, but only data.)
Program to calculate results
The cat_eval program calculates
all of the results for the three sub-tasks (even though the official
measure for the triage subtask is normalized utility and for the
hierarchy annotation subtasks is the F-score). There is a generic
version along with a Windows version as an executable. Python 2.3
or above must be installed to run the program. The program has
been tested on Windows and Solaris but
should run on just about any OS where Python can be installed.
Similar to trec_eval, there is very little error-checking for proper
data format, so data files should be in the format specified
on this page. For any issues, email
Ravi Teja Bhupatiraju at bhupatir@ohsu.edu.
Usage: cat_eval.py data_file gold_standard_file
The package comes with three sets of sample files for each of
subtask. The files annhi.txt and annhiev.txt are slightly
modified versions of pgd+train.txt and pgde+train.txt respectively. The
file retrieved.txt is from an actual run.
These are the files for the triage subtask:
Sample Data: retrieved.txt
Gold Standard: triage+train.txt
These are the files for the annotation hierarchy subtask:
Sample Data: annhi.txt
Gold Standard: pgd+train.txt
These are the files for the annotation hierarchy plus evidence subtask:
Sample Data: annhiev.txt
Gold Standard: pgde+train.txt
Here are the formulae used in the program:
Precision = TP / (TP + FP)
Recall = TP / AP
F-score = 2.0 * precision * recall / (precision + recall)
UtilityFactor = 20
Raw Utility = (UtilityFactor * TP) - FP
Max Utility = (UtilityFactor * AP)
Normalized Utility = (UtilityFactor * TP - FP) / (UtilityFactor * AP)
Here is sample output you should get from the triage subtask sample
data:
>> cat_eval.py retrieved.txt triage+train.txt
Run: OHSU-TRIAGE-CLASSIFIER-V72
Counts: tp=321; fp=1558; fn=54;
Precision: 0.1708
Recall: 0.8560
F-score: 0.2848
Utility Factor: 20
Raw Utility: 4862
Max Utility: 7500
Normalized Utility: 0.6483
To generate results that are amenable to processing by spreadsheets,
statistical programs, and so forth, the command-line tag csv can be added to
generate a space-delimited file:
bash-2.05$ ./cat_eval.py retrieved.txt triage+train.txt csv
Run TP FP FN Precision Recall F-Score Utility Factor Raw Utility Max Utility Normalized Utility
OHSU-TRIAGE-CLASSIFIER-V72 321 1558 54 0.1708 0.8560 0.2848 20 4862.0 7500.0 0.6483